
Imagine you are running a clinical trial. A participant calls to say they have a splitting headache. You treat it, they go home, and life goes on. Now imagine another participant is rushed to the hospital because their heart stopped beating for ten seconds. Both are medical occurrences. Both are adverse events. But only one triggers an emergency alarm that rings across regulatory agencies worldwide.
The difference between these two scenarios isn't just about how bad the patient felt. It is about the outcome. This distinction determines whether you file a routine form next month or send an urgent report within 24 hours. Getting this wrong can delay drug approvals, waste millions in compliance costs, or worse, put patients at risk by hiding critical safety signals under a mountain of noise.
Defining the Core Concepts
To navigate safety monitoring effectively, you must first separate two terms that sound identical but mean completely different things: severity and seriousness. This confusion is the root cause of most reporting errors in clinical research.
Severity describes the intensity of a symptom. A headache can be mild, moderate, or severe. If the pain is blinding and stops you from working, it is a severe headache. However, if it does not require hospitalization and resolves with over-the-counter medication, it remains a non-serious adverse event.
Seriousness, on the other hand, is defined by specific outcome criteria. An event is serious based on what happened to the patient, not how intense the symptom was. According to the International Council for Harmonisation (ICH) E2A guideline, an event is serious if it meets any of six specific outcomes. The intensity of the symptom is irrelevant to this classification.
Dr. Robert Temple, former FDA Deputy Center Director for Clinical Science, noted that confusing these two concepts leads to both over-reporting of trivial issues and under-reporting of critical dangers. When you classify a 'severe' rash as 'serious' simply because it looks bad, you dilute the data. Regulators then struggle to find the actual life-threatening risks hidden in the paperwork.
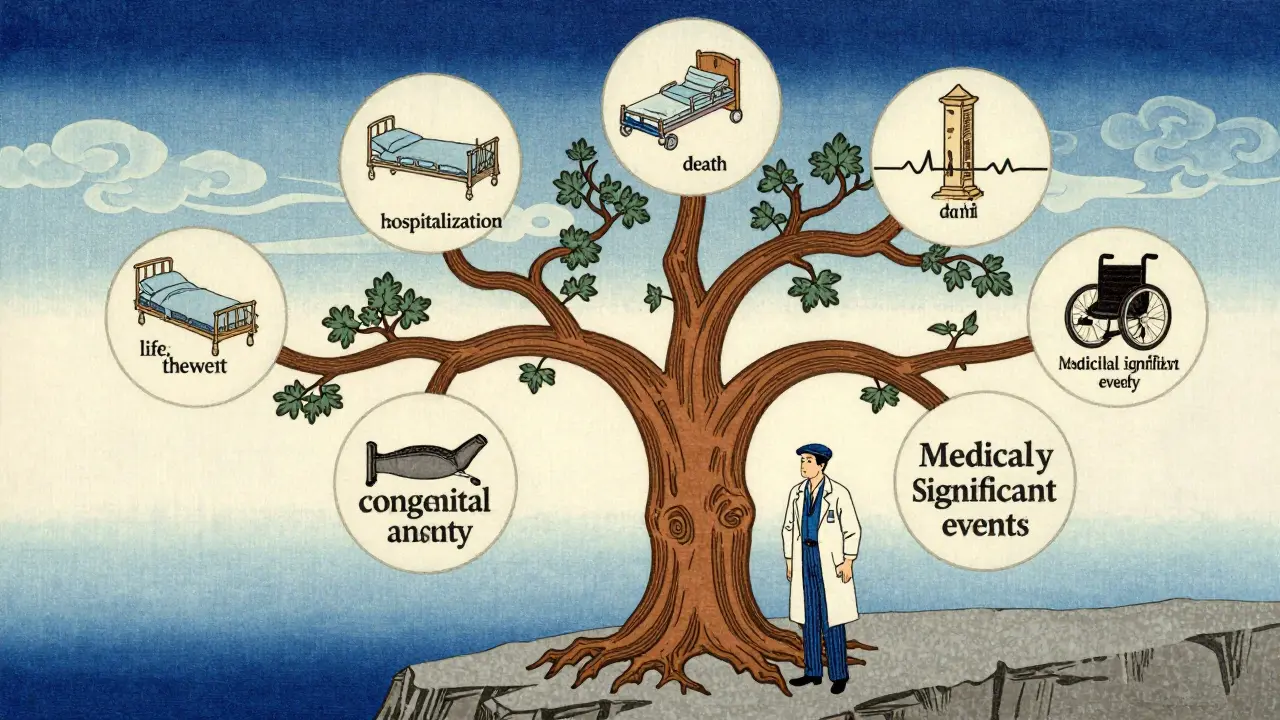
The Six Criteria for Serious Adverse Events
How do you decide if an event is serious? You do not guess. You check against the standard list established by the U.S. Food and Drug Administration (FDA) and adopted globally through ICH guidelines. An adverse event is classified as serious (SAE) if it results in any of the following:
- Death: The patient dies during the study period.
- Life-threatening: The event places the patient at immediate risk of death at the time it occurred. This does not mean the patient actually died, but that if left untreated, death would have been likely.
- Hospitalization: The event requires inpatient admission or prolongs an existing hospital stay. Note that planned surgeries unrelated to the drug are generally excluded unless complications arise.
- Disability: The event causes persistent or significant disability or incapacity. This means a substantial disruption in the ability to carry out normal life functions.
- Congenital Anomaly: The event results in a birth defect or congenital anomaly in a newborn.
- Medically Significant Event: The event may not be immediately life-threatening but requires intervention to prevent one of the above outcomes. Examples include allergic bronchospasm requiring intensive treatment or seizures causing status epilepticus.
If an event meets none of these criteria, it is non-serious. It might be annoying, painful, or even severe in intensity, but it is not serious by regulatory definition.
| Feature | Serious Adverse Event (SAE) | Non-Serious Adverse Event |
|---|---|---|
| Definition Basis | Outcome-based (death, hospitalization, etc.) | Intensity-based (mild, moderate, severe) |
| Reporting Timeline | Expedited (within 24 hours to sponsor) | Routine (per protocol schedule) |
| Regulatory Impact | Triggers expedited reports to FDA/EMA | Included in annual safety summaries |
| Example | Mild fever leading to sepsis and ICU admission | Severe migraine treated with oral meds at home |
Reporting Timelines: The Clock Starts Immediately
Once you determine an event is serious, speed becomes your primary obligation. The regulatory framework mandates strict deadlines to ensure that safety signals reach decision-makers before more patients are harmed.
According to 21 CFR 312.64(b), investigators must report serious adverse events to the sponsor immediately. In regulatory language, 'immediately' means within 24 hours of the investigator learning about the event. This clock starts the moment you become aware of the SAE, regardless of whether you believe the investigational product caused it.
The National Institutes of Health (NIH) National Institute on Aging (NIA) reinforces this, stating that investigators must report to the sponsor within 24 hours, whether or not the event is considered drug-related. Causality is a separate determination made later; it does not pause the reporting clock.
For non-serious adverse events, there is no rush. These are documented in Case Report Forms (CRFs) and reported according to the protocol-specified timetable. This often happens monthly or quarterly during routine data reviews. The goal here is comprehensive data collection, not emergency response.
Who Needs to Know?
Reporting is not a one-way street. Different stakeholders receive different levels of information based on their role in patient safety.
The Sponsor: As mentioned, the sponsor receives the initial notification within 24 hours. They are responsible for evaluating the event and deciding if further regulatory action is needed.
The Institutional Review Board (IRB): In the United States, IRBs protect human subjects. The University of Michigan’s Research A-Z guidance specifies that serious adverse events must be reported to the IRB within 7 days. Non-serious events may only be reported during the next scheduled continuing review, or not at all, depending on the protocol.
Regulatory Authorities: The FDA and European Medicines Agency (EMA) require sponsors to submit expedited safety reports. For life-threatening events, the FDA mandates submission within 7 calendar days. For other serious, unexpected adverse reactions, the deadline is 15 calendar days.

Common Pitfalls in Classification
Even experienced researchers stumble on this topic. Why? Because real-life medical situations are messy, and definitions can feel rigid. Here are the most common traps:
Emergency Room Visits: Does a trip to the ER automatically make an event serious? Not necessarily. If a patient visits the ER for severe anxiety but is discharged without admission and suffers no permanent damage, it is often non-serious. The NIH clarified in 2023 that ER treatment alone qualifies as serious only if it meets other criteria, such as being life-threatening. Many sites incorrectly flag all ER visits as SAEs, creating unnecessary administrative burden.
Psychiatric Events: Mental health conditions pose unique challenges. A patient experiencing 'severe depression' has a high-intensity symptom. Unless this leads to hospitalization, suicide attempt (life-threatening), or significant disability, it may remain a non-serious adverse event graded as severe. However, due to the inherent risk, many protocols require heightened scrutiny for psychiatric AEs regardless of classification.
Baseline Conditions: In oncology trials, patients often enter with poor health. A cancer patient who develops pneumonia might have a condition that is expected given their disease state. Determining if this is an SAE related to the drug or part of the natural disease progression requires careful judgment. The SWOG Cancer Research Network found that nearly 32% of SAE reports required amendment due to incorrect seriousness classification, often stemming from these baseline complexities.
Best Practices for Accurate Reporting
To avoid costly errors and ensure patient safety, implement these systematic approaches in your trial operations:
- Use Decision Trees: Adopt the NIA’s four-question decision tree for every event. Did it result in death? Was it life-threatening? Did it require hospitalization? Did it cause disability? If yes to any, it is serious. If no, it is not.
- Train Your Team Annually: Knowledge fades. The Association of Clinical Research Professionals reports that top institutions require annual refresher training on seriousness criteria. Ensure everyone from coordinators to principal investigators understands the difference between severity and seriousness.
- Leverage Technology: Automated tools are improving rapidly. AI systems now classify seriousness with nearly 90% accuracy, compared to 76% for human reviewers alone. Use electronic data capture systems with built-in logic checks to flag potential SAEs before submission.
- Document Everything: Keep clear records of why an event was classified as non-serious if it involved an ER visit or intense symptoms. This documentation protects you during audits and helps regulators understand your rationale.
- Consult Early: If you are unsure, contact the sponsor’s medical monitor or your IRB office before filing. It is better to ask questions than to submit incorrect reports that trigger investigations.
The global market for clinical trial safety management is growing, projected to reach $5.89 billion by 2028. Much of this growth is driven by the need for better, faster, and more accurate adverse event processing. By mastering the distinction between serious and non-serious events, you contribute to a system that prioritizes genuine safety signals over administrative noise.
Is a severe headache considered a serious adverse event?
No. Severity refers to the intensity of the symptom, while seriousness refers to the outcome. A severe headache is only serious if it leads to one of the six specific outcomes, such as hospitalization, life-threatening status, or significant disability. If the headache is intense but resolved with medication at home, it is a non-serious adverse event, even if graded as severe.
What is the deadline for reporting a serious adverse event to the sponsor?
Investigators must report serious adverse events to the sponsor within 24 hours of becoming aware of the event. This timeline applies regardless of whether the event is believed to be related to the investigational product. The clock starts immediately upon discovery.
Does a visit to the emergency room always constitute a serious adverse event?
Not necessarily. An ER visit is only serious if it meets other criteria, such as being life-threatening or resulting in hospitalization. If a patient goes to the ER for evaluation but is discharged without admission and suffers no permanent harm, the event is typically classified as non-serious, though it should still be documented carefully.
How do non-serious adverse events differ from serious ones in reporting?
Non-serious adverse events are reported routinely according to the study protocol, often monthly or quarterly via Case Report Forms. They do not trigger expedited notifications to regulators or IRBs. Serious adverse events require immediate notification (within 24 hours) to the sponsor and potentially within 7-15 days to regulatory authorities like the FDA.
What are the six criteria that define a serious adverse event?
The six criteria are: death, life-threatening occurrence, inpatient hospitalization or prolongation of existing hospitalization, persistent or significant disability/incapacity, congenital anomaly/birth defect, and a medically significant event that requires intervention to prevent permanent impairment. Meeting any one of these makes an event serious.